Indice [nascondi]
Che cos’è l’amiloidosi?
L’amiloidosi rappresenta un gruppo eterogeneo di patologie caratterizzate dal deposito extracellulare di proteine fibrillari insolubili.
In condizioni fisiologiche, le proteine prodotte dal nostro organismo si ripiegano naturalmente in una forma tale da consentire il loro specifico funzionamento.
In caso di mal ripiegamento invece, l’organismo cerca di individuare e rimuovere le proteine anomale: se il tentativo di disgregazione fallisce o la produzione è eccessiva le proteine alterate iniziano a legarsi tra loro per formare fibre lineari e rigide (le fibrille) che a loro volta si accumulano in organi e tessuti.
Eziopatogenesi
Le forme più comuni di amiloidosi sono:
- La AL o primaria, dovuta al deposito di catene leggere libere monoclonali. Una tipologia di globuli bianchi chiamata plasmacellule, ha la funzione di produrre immunoglobuline, ossia anticorpi costituiti da catene leggere e pesanti. Una produzione eccessiva di catene leggere mal ripiegate è responsabile di questa forma di amiloidosi che è la più comunemente diagnosticata e quella a prognosi peggiore.
- L’amiloidosi correlata al deposito di transtiretina mutata (ATTRm), forma ereditaria della patologia. Esistono oltre 100 mutazioni note di TTR responsabili della sua instabilità e del mal ripiegamento. Inoltre, le diverse mutazioni sono correlate a diversi organi bersaglio a livello dei quali la malattia tende a manifestarsi clinicamente. Si pensa che l’amiloidosi da TTR sia più frequente dell’amiloidosi AL, anche se spesso non viene riconosciuta e diagnosticata.
- L’amiloidosi sistemica senile (TTR wt o SSA), ad insorgenza tardiva, correlata alla deposizione di transtiretina non mutata (wild type).
- L’amiloidosi Aβ2 M, che si presenta in pazienti con insufficienza renale avanzata e dializzati da molti anni. Una proteina serica circolante, la beta-2 microglobulina, non riuscendo ad attraversare il filtro della dialisi, non può essere smaltita e l’amiloide risultante dalla sua aggregazione si accumula nei tessuti, specialmente nelle articolazioni e nei tendini.
- L’’amiloidosi AA, una forma più rara legata all’accumulo della proteina siero amiloide A circolante che si manifesta secondariamente a condizioni infiammatorie croniche ed infezioni. Tra queste ricordiamo disturbi reumatici, malattie infiammatorie croniche intestinali, tubercolosi, osteomielite, lupus eritematoso sistemico e sindromi febbrili ereditarie come la febbre mediterranea familiare.
Sintomi e segni clinici
I sintomi dell’amiloidosi sono estremamente variabili in base ai tessuti colpiti, ma le manifestazioni cardiache sono esclusive delle forme AL e ATTR ( sia mutata che wild type).
Possiamo riscontrare disturbi del ritmo, cardiomiopatia ipertrofica o restrittiva con successiva evoluzione in scompenso cardiaco conclamato, insufficienza renale (oliguria, proteinuria, edemi periferici), alterazioni gastrointestinali (nausea, diarrea alternata a stipsi, perdita di peso, inappetenza), neuropatia amiloide (alterato controllo degli sfinteri, sudorazione eccessiva, formicolio degli arti superiori ed inferiori, sindrome del tunnel carpale, decadimento cognitivo), difficoltà nella deglutizione e macroglossia.
Iter diagnostico
Un’accurata diagnosi è fondamentale per stabilire la terapia più adeguata, la quale varia in base al meccanismo patogenetico coinvolto.
Purtroppo a causa dei sintomi aspecifici e difficili da riconoscere, il ritardo nella diagnosi è ancora un evento comune.
L’iter diagnostico prevede:
-
- raccolta di informazioni riguardanti la storia clinica familiare e personale del paziente;
- esami di laboratorio volti alla ricerca della proteina anomala su sangue e urine;
- biopsia con agoaspirato ed esame istologico, la conferma diagnostica consiste nel riconoscimento della sostanza amiloide mediante analisi di un campione di tessuto, generalmente il tessuto adiposo addominale. Se il grasso addominale aspirato è negativo all’analisi istologica ma il sospetto clinico della malattia è elevato, è indicato effettuare una biopsia dell’organo presumibilmente coinvolto (ad es. reni, fegato, cuore);
- analisi genetiche qualora si sospetti una forma ereditaria;
- esami strumentali per quantificare il danno d’organo (risonanza magnetica, scintigrafia, PET – tomografia ad emissione di positroni).
Ricordiamo infine, gli esami strumentali cardiologici che ci orientano verso la diagnosi in caso di sospetto di amiloidosi cardiaca:
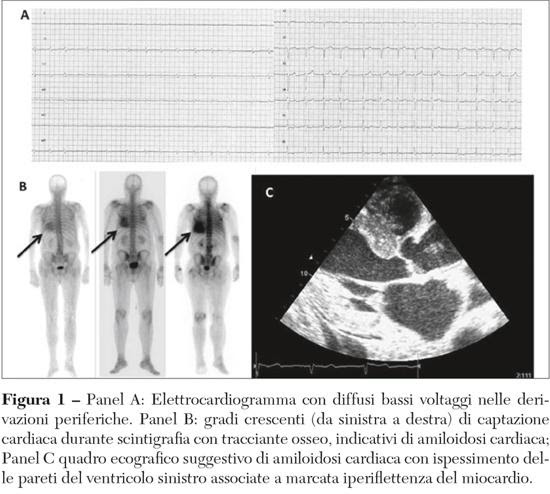
- l’elettrocardiogramma può rilevare la presenza di bassi voltaggi e di un pattern di “pseudonecrosi”, dovuto alla capacità isolante della sostanza amiloide;
- l’ecocardiogramma mette in evidenza alcuni segni suggestivi della malattia come l’ispessimento del setto interatriale e del setto interventricolare, l’aspetto “brillante e granulare” del miocardio, l’ispessimento dei lembi valvolari, una lieve falda di versamento pericardico;
- la scintigrafia con bifosfonati, il cui razionale è basato sulla capacità di questi ultimi di legare la sostanza amiloide accumulata. Con questa tecnica è possibile identificare il deposito di tipo TTR e non AL, facilitando nella pratica clinica la diagnosi differenziale tre le due forme della patologia;
- la risonanza magnetica cardiaca, metodica sensibile nell’individuare l’ispessimento delle pareti cardiache da accumulo di sostanza amiloide.
Strategie terapeutiche
L’attuale strategia terapeutica dell’amiloidosi è di tipo eziologico ed ha lo scopo di arrestare la progressione del danno d’organo.
Nell’amiloidosi primaria (AL) il trattamento consiste nella chemioterapia, con lo scopo di interrompere la crescita di plasmacellule che producono le catene leggere anticorpali anomale.
Nella forma infiammatoria (AA) la terapia è mirata a trattare l’infezione o la condizione infiammatoria cronica per rallentare l’accumulo progressivo di amiloide, riducendo i livelli della proteina siero amiloide A circolante.
In caso di amiloidosi ereditaria il trapianto di fegato, sede della sintesi della proteina mutata, permetterebbe la produzione di TTR normale. I periodi di attesa per la donazione di organi sono generalmente lunghi, ma il trapianto è un’opzione possibile per quei pazienti la cui malattia non sia in stadio troppo avanzato.
Due opzioni farmacologiche in grado di prevenire il mal ripiegamento della TTR mutante in amiloide sono il diflunisal e il tafamidis, piccole molecole che si legano alle proteine precursore e stabilizzano la loro struttura in modo da impedire la formazione delle fibrille. Questi farmaci possono essere utilizzati anche nella forma senile di amiloidosi.
Da pochi mesi in Italia è inoltre disponibile un nuovo farmaco, il Patirisan, approvato per il trattamento di pazienti adulti affetti da amiloidosi ereditaria in presenza di polineuropatia.
Il suo meccanismo d’azione è basato sul “silenziamento” di uno specifico RNA per “spegnere” la produzione di transtiretina anomala nel fegato e rappresenta una delle frontiere più promettenti nel trattamento della patologia.
Nell’amiloidosi Aβ2 M il trapianto di rene è considerata l’opzione terapeutica migliore.
Ricordiamo infine l’importanza di affiancare alla terapia eziologica una terapia di supporto con lo scopo di promuovere la funzione degli organi colpiti e migliorare la qualità di vita dei pazienti (terapia di supporto cardiologica, nefrologica, neurologica, terapia del dolore e supporto nutrizionale).
Contatta l’esperto in merito a questo argomento.
Dott. Federica Verrillo
Cardiologo in Formazione
Dipartimento di Scienze Mediche Traslazionali
Malattie dell’Apparato Cardiovascolare
Università della Campania Luigi Vanvitelli
AORN Monaldi Napoli – AORN Sant’Anna e San Sebastiano Caserta
Revisione a cura del: Prof. Giuseppe Limongelli
Professore associato
Unità di Malattie genetiche e Rare Cardiovascolari
Università della Campania “Luigi Vanvitelli”,
AORN Colli, Ospedale Monaldi, Napoli, Italia
Institute of Cardiovascular Sciences,
University College of London, London, UK